Notes from MCB 104 Pt.2 Genomics Lectures
High-level Concepts
Sequencing techniques
Genome sequencing
- Fragment DNA using
- Restriction enzyme digestion (e.g. EcoR1)
- If a restriction enzyme recognizes a greater # of bases, it’s harder to find matches in the sequence, and they generate larger fragments (travel less distance in gel electrophoresis)
- Sonication
- More sonication cycles gives us smaller fragments
- Then do gel electrophoresis
- Restriction enzyme digestion (e.g. EcoR1)
- Clone DNA fragments into plasmid vector backbone (could also use PCR to amplify sequence)
- Creates a library of plasmids
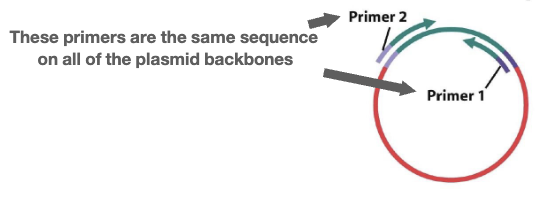
- Sequence the entire library using paired-end sequencing: sequences of both ends of a piece of DNA
- Primers are used on both ends, with our DNA fragment in the middle; the primers are the same sequence on all the plasmid backbones
- Used to assemble contigs (see next step)
- Sequencing process: when ddNTP (fluorescent base) added, it terminates the read, so the reads are different lengths ????
- DNA to sequence → denature DNA (into single strands) and anneal primer ⇒ primer extension to copy and label DNA: DNA pol, nucleotides ATCG extend the primer, and ddNTPs terminate the extension
- Detect the sequence on a gel to separate fragments based on size; could verify the efficiency of digestion/sonication, and extract DNA fragments of a specific size for further cloning/sequencing/fragmenting
- Primers are used on both ends, with our DNA fragment in the middle; the primers are the same sequence on all the plasmid backbones
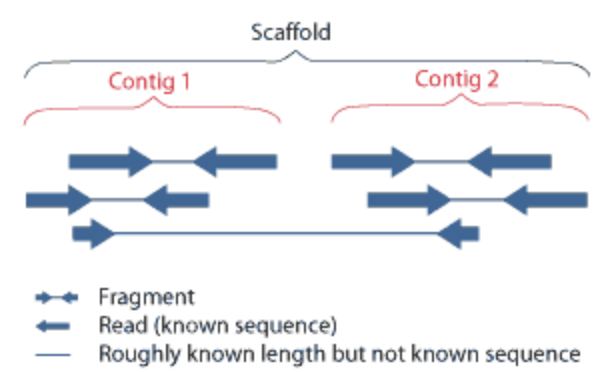
- Align overlapping paired-end reads to make contigs: a set of overlapping DNA segments/sequences that overlap in a way to provide a contiguous representation of a genomic region
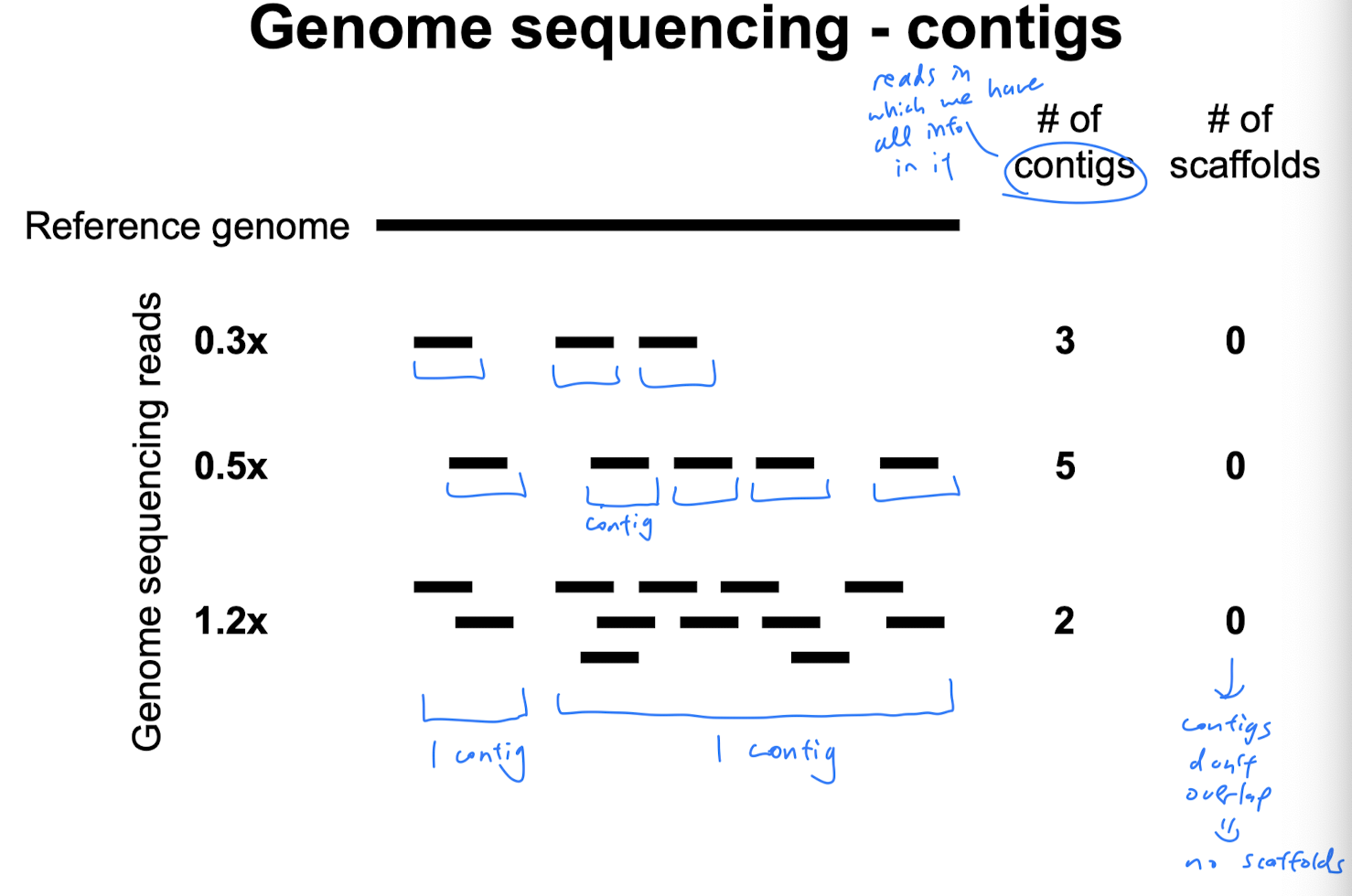
- After reaching 1x coverage, if we keep increasing coverage, then the number of contigs decreases because we get more overlapping regions, so the contigs assemble into longer ones, but there are fewer distinct contigs
- Before reaching 1x coverage, if we keep increasing coverage, the # of contigs increases
- Use paired-end information to group contigs into scaffolds (overlapping contigs create scaffolds): large stretch of sequence built from multiple contigs; may contain gaps
Relationship between scaffolds and contigs
RNA-Seq
High-throughput shotgun cDNA sequencing
- Purpose: to quantify gene expression by sequencing RNA present in a sample; used to identify which parts of a genome are being actively transcribed into RNA
- RNA-Seq also allows for
- Locations of genes and exons
- Can reveal coding sequence polymorphism
Steps
- Harvest mRNA - we will only be able to see mRNA transcripts and cDNA library from the specific cell type we’re isolating the mRNA from
- Reverse transcribe mRNA to cDNA using reverse transcriptase
- Also use DNA polymerase to make single-stranded cDNA → double-stranded cDNA
- Clone cDNA into vectors
- Sequence the 5’ and 3’ ends of each cDNA insert/clone/plasmid (aka paired-end reads) to generate Expressed Sequence Tags (ESTs): a short sub-sequence of cDNA transcript sequence
- cDNA and ESTs reveal exons (no introns!) and transcribed portions of the genome
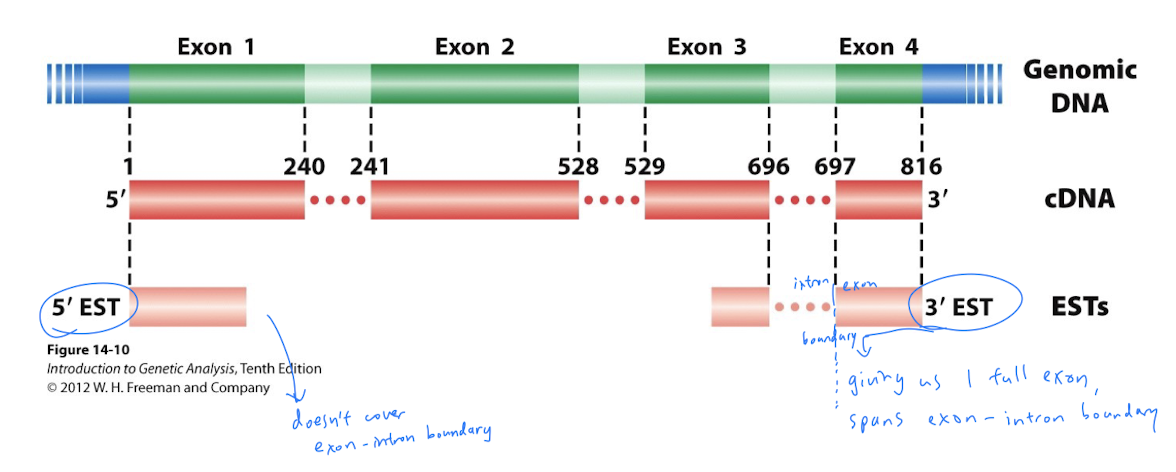
- cDNA and ESTs reveal exons (no introns!) and transcribed portions of the genome
- Align reads to assemble genome
Relationship between genomic DNA, mRNA, cDNA, and ESTs
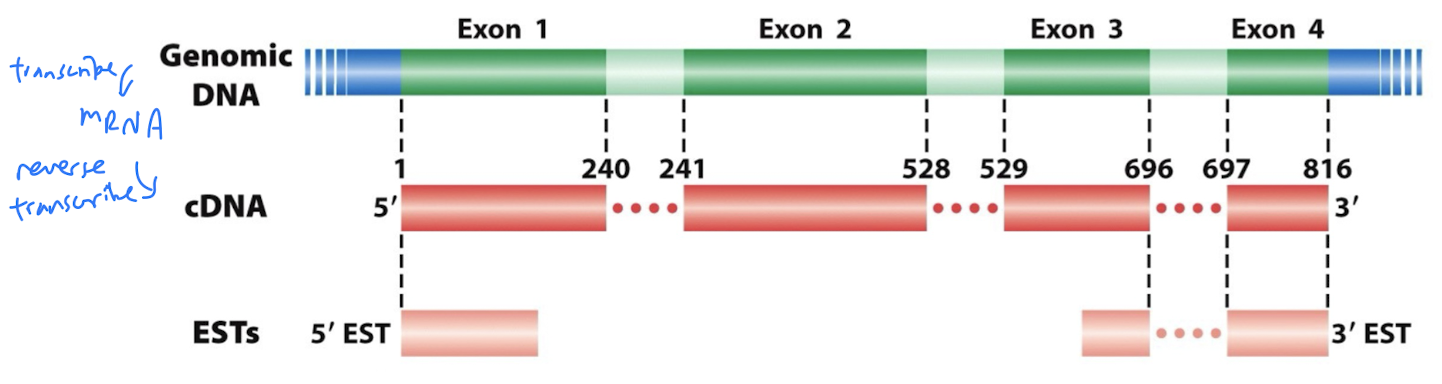 Genomic DNA is transcribed into mRNA
mRNA is reverse transcribed into cDNA
ESTs are a sub-sequence of cDNA
Genomic DNA is transcribed into mRNA
mRNA is reverse transcribed into cDNA
ESTs are a sub-sequence of cDNA
ATAC-Seq (Assay for Transposase-Accessible Chromatin)
- Purpose: determine chromatin accessibility across genome by sequencing regions of open chromatin
- We can use this information to do further testing to reveal putative enhancer regions using ChIP-Seq, for e.g.
Steps
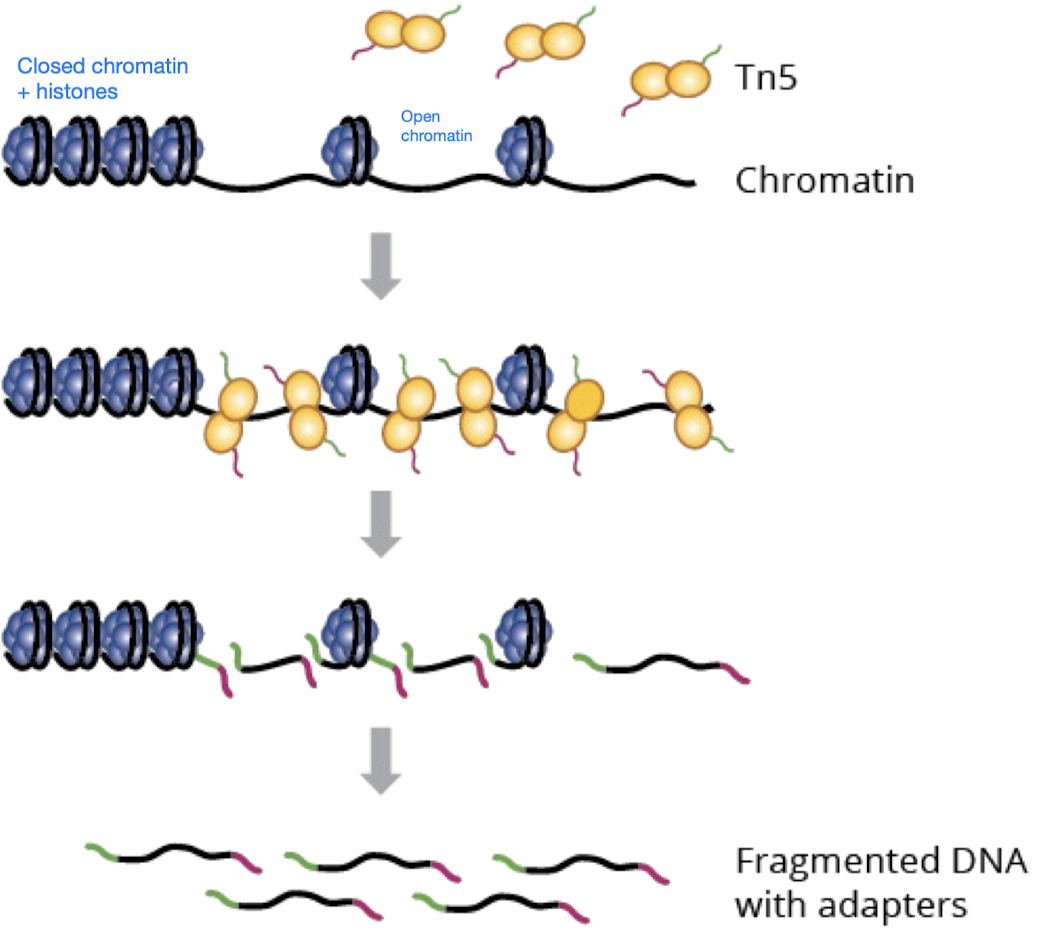
- Tn5 transposase (simultaneously fragments chromatin regions and tags the regions with sequencing adapters) performs tagmentation on open chromatin
- Tn5 is a transposase, so it cuts and pastes (vs. restriction enzyme, which only cuts but doesn’t paste)
- Tn5 attaches sequencing adapters so the DNA fragments can be recognized later
- Amplify fragmented DNA with adapters using PCR
- Use paired-end sequencing to align the fragments to the genome to find out where Tn5 cut, revealing where chromatin is accessible
ChIP-Seq (Chromatin Immunoprecipitation)
- Purpose: identifies binding sites** (e.g. enhancer region) of DNA-associated proteins (e.g. transcription factors) and can be used to map global binding sites for a given protein
- Can also tell us the identity of the protein (e.g. tf, histone) binding to the DNA region (bc we have to use a protein-specific antibody)
Steps
- Crosslink DNA and proteins (TFs) using formaldehyde - to secure the interactions between the DNA and the TFs, to make sure they’re linked for all the following steps
- Sonicate (fragment) DNA
- Add antibody specific to protein (TF) and co-immunoprecipitate the TF-bound DNA using the TF-specific antibody
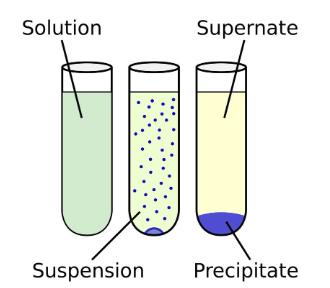
- The mixture would look like:
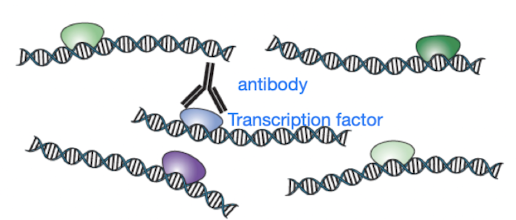
- The co-immunoprecipitate includes 3 molecules:
- Antibody that is specific to the target protein, i.e. the antibody only recognizes the target protein (e.g. TF) linked to DNA
- The target protein (e.g. a TF)
- DNA (that the target protein is bound to; remember, target protein and DNA were crosslinked in the first step!)
- The co-immunoprecipitate is precipitated out, and we can dump the supernatant
- The mixture would look like:
- Purify the DNA - isolate the DNA and dissociate the TF and antibodies
- Sequence the DNA - purpose of ChIP-Seq is to identify binding sites of DNA-associated proteins (e.g. TFs, histones), so sequencing DNA fragments
Genome arrangement
2 main mechanisms that lead to chromosomal rearrangements
- DNA DSBs (double-strand breaks)
- Repeated sequences and transposons Ways chromosomes interact with each other
- On 1 chromsome
- Between homologous chromosomes
- Between non-homologous chromosomes
Gain of genetic material
- Duplication
- DSB - Between 2 homologous chromosomes - 2 DNA DSBs, but the ends of 1 strand of a chromosome fuse with the other strand of chromosome instead of with itself - leads to simultaneous deletion and duplication
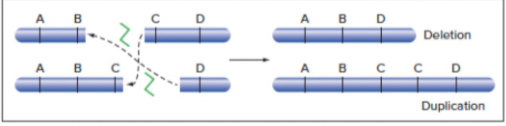
- Repeated sequences in homologous chromosomes - simultaneous deletion and duplication
- Crossing over in wrong site in chromosome (i.e. if there is another site in the chromosome that happens to have the same sequence)
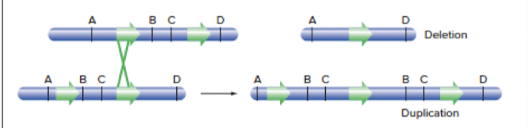
- Crossing over in wrong site in chromosome (i.e. if there is another site in the chromosome that happens to have the same sequence)
- DSB - Between 2 homologous chromosomes - 2 DNA DSBs, but the ends of 1 strand of a chromosome fuse with the other strand of chromosome instead of with itself - leads to simultaneous deletion and duplication
- Extra chromosome
Loss of genetic material
- Deletion
- DNA DSBs
- On 1 chromosome
- 1 DNA DSB: repairing the DSB via NHEJ (fusing both ends of break while omitting the segment in the middle) leads to deletion
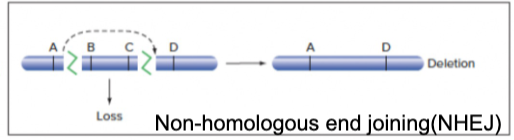
- 1 DNA DSB: repairing the DSB via NHEJ (fusing both ends of break while omitting the segment in the middle) leads to deletion
- Between 2 homologous chromosomes - 2 DNA DSBs, but the ends of 1 strand of a chromosome fuse with the other strand of chromosome instead of with itself - leads to simultaneous deletion and duplication
- On 1 chromosome
- Repeated sequences
- in homologous chromosomes - simultaneous deletion and duplication
- Crossing over in wrong site in chromosome (i.e. if there is another site in the chromosome that happens to have the same sequence)
- Crossing over in wrong site in chromosome (i.e. if there is another site in the chromosome that happens to have the same sequence)
- in 1 chromosome: aberrant crossover event leads to loss of genetic material
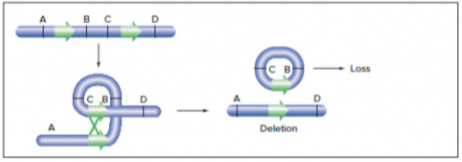
- in homologous chromosomes - simultaneous deletion and duplication
- DNA DSBs
Relocation of genetic material
- Translocation
- DNA DSB: Between 2 nonhomologous chromosomes: both strands have DNA DSBs, and fuse with each other
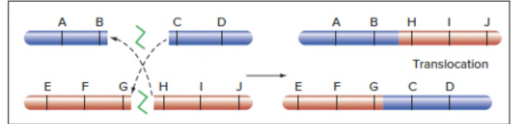
- Repeated sequences: crossing over between 2 nonhomologous chromosomes
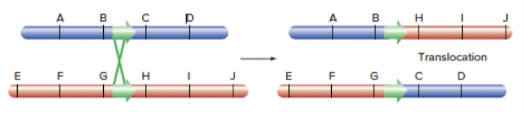
- DNA DSB: Between 2 nonhomologous chromosomes: both strands have DNA DSBs, and fuse with each other
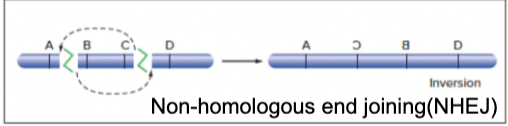
- Inversion
- DNA DSB: on 1 chromosome, repairing the DSB via NHEJ, but the segment in the middle generated by the DSB gets inverted in the process of repair
- Repeated sequences: looping event creates inversion
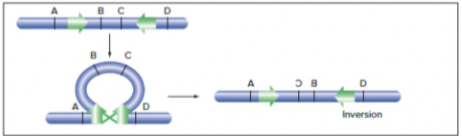
- DNA DSB: on 1 chromosome, repairing the DSB via NHEJ, but the segment in the middle generated by the DSB gets inverted in the process of repair
Transposons
2 classes Like repeated sequences, can lead to chromosomal rearrangements
1. Retrotransposons (copy and paste)
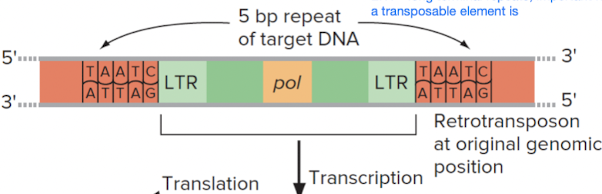
- Moves (copies) DNA via reverse transcription into an RNA intermediate
- Is flanked by long terminal repeats (LTRs) on left and right ends
- Requires reverse transcriptase to produce RNA intermediate
Steps
- Retrotransposon (DNA) gets transcribed, giving us mRNA intermediate
- mRNA intermediate is reverse transcribed by reverse transcriptase; we obtain ds-cDNA copy of retrotransposon
- Meanwhile, endonuclease cleaves the identifying site in the new location in genome where retrotransposon is supposed to be inserted
- Insertion of ds-cDNA into new site in genome
2. DNA Transposon (cut and paste)
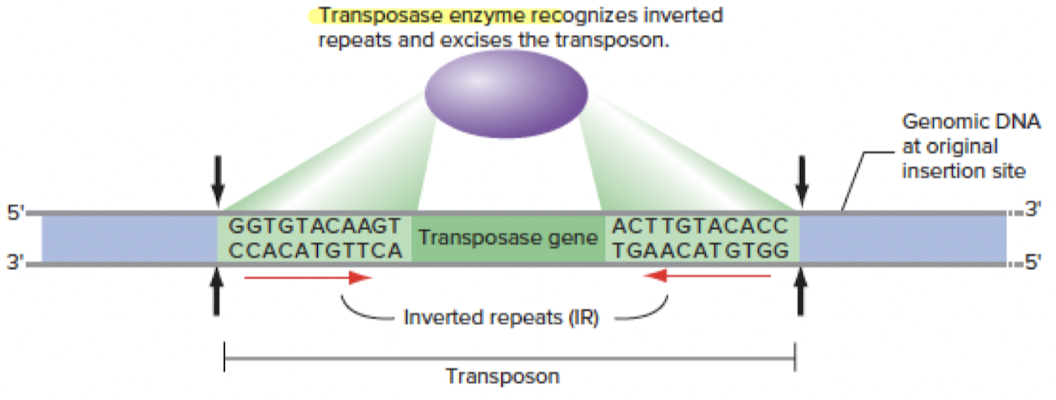
- Moves DNA directly (cuts and pastes DNA)
- Is flanked by inverted repeats (IRs) on left and right ends
- Requires transposase to recognize IRs and excise the transposon
Steps
- Transposase recognizes IRs to locate the DNA transposon to know where to cut
- Transposase cuts the DNA transposon, and the DNA transposon
- can stay in place
- or can be inserted into new site in genome (and OG position can be repaired by copying homologous chromosome
Promoters
- Def: region of DNA upstream of a gene where relevant proteins (e.g. RNA pol, transcription factors) bind to to initiate transcription of that gene; resulting transcription produces an RNA molecule
- Location: 5’ adjacent (upstream) to gene’s transcription start site, on the same chromosome (aka “cis-acting”)
Enhancers
aka “regulatory DNA”
- Def: region of DNA that positively regulates transcription, often in a spatial and/or temporal specific manner (via differential accessibility and transcription factor binding, also combinatorial logic of multiple enhancers)
- Transcription factors (e.g. activators and repressors are classes of tfs) bind to enhancers to regulate transcription
- Potential locations: anywhere (upstream 5’ or downstream 3’, close to or far away from the gene) as long as its on the same chromosome
Transcription factors regulate enhancers: activators & repressors
- Activators and repressors are categories of transcription factors and are only cell-specific (e.g. activators and repressors specific to the skin or eye)
- Enhancer activity is space-specific and time-specific
Reporter assay
- Purpose: tools to test enhancer activity, to understand gene regulation
- Answers question: Is a specific region of DNA an enhancer or not?
Steps
- Clone putative enhancer (or other regulatory DNA sequence of interest, e.g. promoter) into DNA vector with a general basal promoter (gives basal/minimal level of gene expression) and a reporter gene (e.g. GFP, LacZ) whose product is easy to measure
- Inject recombinant constructs into host embryos (make transgenic by insertion into germ line)
- We assess the enhancer’s (or other regulatory DNA sequence of interest’s) activity by measuring expression level of the reporter gene’s product (e.g. GFP, visualize and quantify gene expression levels using fluorescence microscopy/flow cytometry)
Genetic screens
- Genetic screen = procedure used to create and detect a mutant organism
Complementation Testing
- Complementation: the ability of 2 mutants in combination to restore a normal phenotype (heterozygosity for loss-of-function mutant recessive alleles for 2 different genes produces a normal phenotype)
- Complementation test: a test to determine if 2 mutations…
- complement (are in different genes, so can restore normal phenotype) ”+“
- or don’t complement (are in same gene) ”-” → mutations that don’t complement each other are in the same complementation group
- Saturation mutagenesis: systemic approach for finding all the genes in the genome that can mutate to affect a biological process
- We know a genetic screen is saturated when we stop finding new loci/genes, but continue to find more alleles (mutants) of the same loci; i.e. until all identified complementation groups each have multiple alleles, the screen is not saturated
Example problems
- Example problem 1
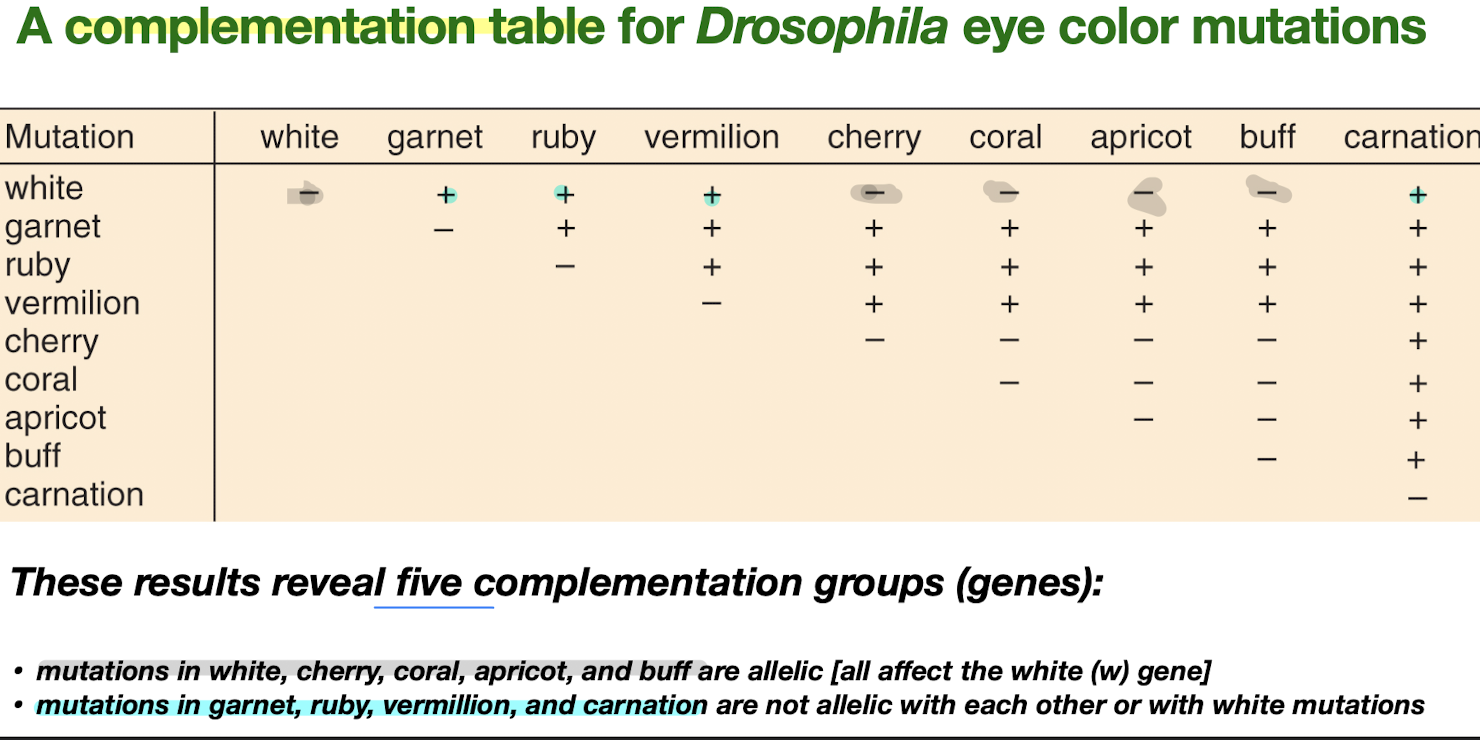
- Ones with (-) in the same row = in the same complementation group
- For each problem, before approaching, identify if a gene is dominant first! If it is, its crosses won’t give us much info about complementation groups
- Example problem 2

Forward Genetics
- Def: start with phenotype of interest, then stimulate random mutagenesis (with e.g. chemicals, x-ray), finally determine which genes are responsible for phenotype of interest
- Application of forward genetic screens in model organisms: they can inform upon human disease, bc when we id key genes and pathways in model organism, it can give us info on conserved functions in human biology
Reverse Genetics
- Def: start with gene of interest, then do targeted mutagenesis (only perturb our gene of interest) via e.g. CRISPR-Cas9, then screen in model organism for what the gene’s phenotype is
- Strategies for reverse genetics
- Acts on RNA (thus makes temporary changes bc only affects gene expression)
- RNAi: manipulates RNA and affects gene expression
- Acts on DNA (thus makes permanent changes in genome)
- Homologous Recombination (HR)
- CRISPR, ZFNs, TALENs - perturb gene of interest, then screen for phenotype
- Acts on RNA (thus makes temporary changes bc only affects gene expression)
Genome editing
Double-Strand Break Repair - 2 repair pathways
- HR (involves strand invasion etc.)
- Involves using homologous sequence (in nature) or using plasmid with homology domains (same sequence as our endogenous gene locus)
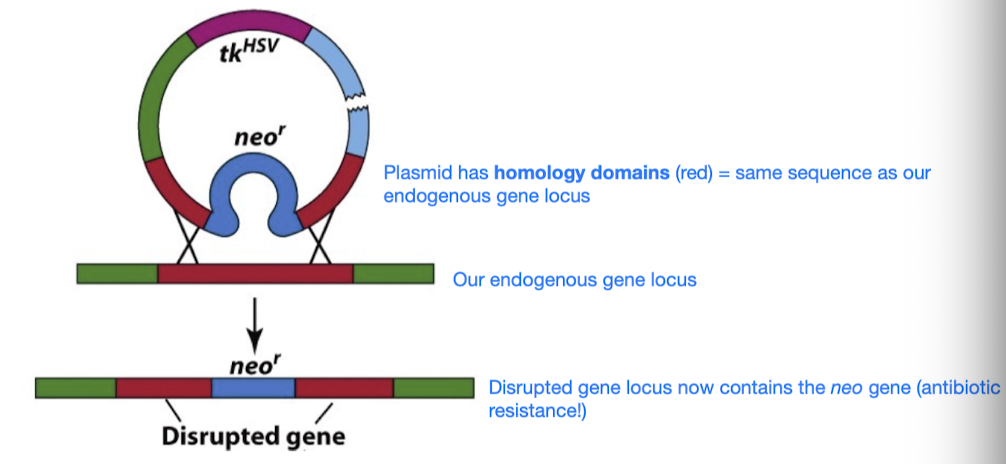
- Involves using homologous sequence (in nature) or using plasmid with homology domains (same sequence as our endogenous gene locus)
- NHEJ (straight up ligate the ends)
- Downside: generates a high frequency of indels at the repair junctions
left off Anki
Programmable Nucleases Allow Genome Engineering
- Zinc Finger Nucleases (ZFNs), TALENs, and CRISPR-Cas9 all have a programmable, DNA-sequence-specific binding domain coupled to an endonuclease activity (i.e. they can all induce DSBs in DNA, leading to alterations in genome thru either repair mechanism, HR or NHEJ)
- Their differences (extra info kinda)
- ZFNs and TALENs: ZFNs and TALENs are proteins that directly bind to DNA sequences
- CRISPR: uses RNA (e.g. gRNA, tracrRNA, crRNA) to direct the Cas9 enzyme to the target DNA sequence
CRISPR-Cas9
- CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) is a segment of DNA containing short repetitions of base sequences (spacers and repeats)
- Origin: found in bacterial immune system
In nature (bacteria)
Components
- CRISPR, a region of DNA which consists of spacers and repeats
- 🦠 Spacers: all different sequences, each serves as a genetic memory of past infections; when prev encountered virus tries to infect the bacterium again, spacers help guide Cas protein to the matching DNA to destroy the invader
- Repeats: short, repetitive, palindromic DNA sequences interspersed between spacers; involved in precr-RNA processing: tracrRNA aligns with repeats, allowing RNase III to cut in correct location
- CRISPR (DNA) is then transcribed into crRNA (CRISPR RNA): contains region complementary to target DNA sequence; crRNA is designed based on DNA sequence we want to edit, and complementarity lets crRNA bind to the target DNA sequence so Cas9 can cut there
- tracrRNA (trans-activating CRISPR RNA): binds to both the crRNA and the Cas9 protein (enzyme), holds the tracrRNA-crRNA-Cas9 complex together
- Involved in pre-crRNA processing (brings Cas9 cleave long strands of pre-crRNA, resulting in release of individual crRNA units)
- Involved in the actual editing of the invader’s genome
- Cas9 gene
- Then transcribed into Cas9 mRNA
- Then translated into Cas9 protein (an endonuclease)
- RNase III: processes pre-crRNA into mature crRNA by cleaving the long strand of pre-crRNA at specific sites, resulting in release of individual crRNA units
Steps
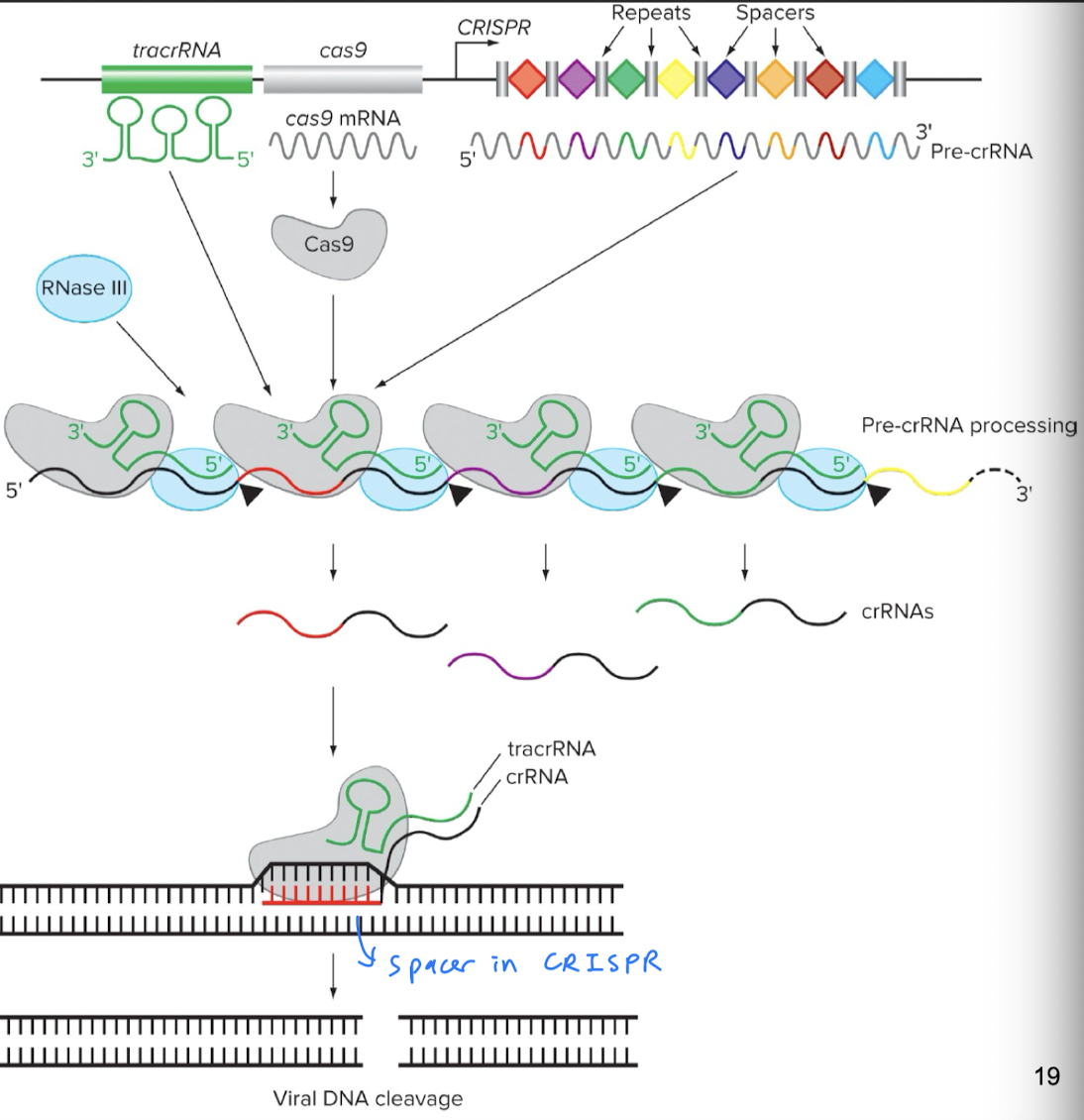
- Spacer acquisition: bacterium is invaded by a virus, and a small piece of the virus’ DNA is captured and incorporated into the CRISPR locus in the bacterium’s genome. This new DNA fragment is called a new spacer
- crRNA biogenesis: CRISPR DNA locus is transcribed into pre-crRNA, which undergoes pre-crRNA processing to turn into short crRNA molecules (1 spacer sequence + partial repeat sequence)
- pre-crRNA processing: involves RNase III, Cas9, and tracrRNA, tracrRNA-Cas9 complex lines up with pre-crRNA, and RNase III cuts the pre-crRNA in the right place
- Formation of surveillance complex: crRNA + Cas9 protein + tracrRNA complex forms. The crRNA is the guide that directs the crRNA-Cas 9 protein-tracrRNA complex to the target DNA sequence matching the crRNA’s spacer sequence.
- Target DNA recognition and cleavage: once target DNA is recognized (using crRNA’s spacer sequence), Cas9 creates a DSB at the target DNA site
- Target DNA repair (for the invader)
In biotech
Components
- Instead of needing CRISPR (the whole DNA region with spacers and repeats) and processing pre-crRNA to crRNA, we can generate crRNA directly
- Instead of having 2 separate molecules, crRNA and tracrRNA, we can create sgRNA (single-guide RNA) containing crRNA + linker loop + tracrRNA
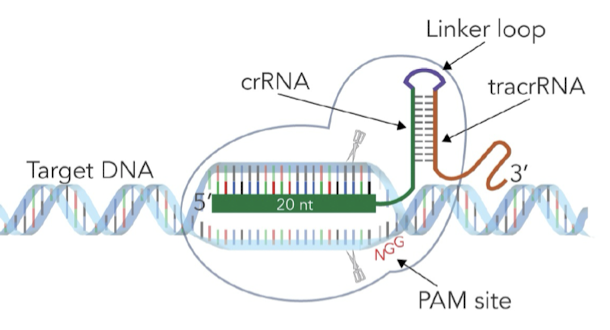
- Purpose of linker loop: stabilizes tracrRNA to crRNA to provide more efficient editing
Steps (in biotech)
- Design sgRNA: sgRNA is complementary to the specific target sequence in genome we want to edit and also includes a sequence that binds to the Cas9 enzyme
- Formation of CRISPR-Cas9 complex (sgRNA + Cas9 protein): sgRNA and Cas9 nuclease are introduced into the cell. Inside the cell, sgRNA binds to Cas9 protein.
- Target DNA recognition and cleavage: CRISPR-Cas9 complex finds sequence in DNA that matches the sgRNA. sgRNA base pairs with complementary DNA sequence. Cas9 makes a DSB.
- DNA DSB repair: 2 main pathways for DSB repair
- NHEJ
- HDR
Applications of CRISPR
- Treating disease: target disease loci by either repairing a mutated gene or inactivating a dominant-negative (dominant, disease-causing) allele
- For a genome-wide screen (forward genetic screen): CRISPR libraries of sgRNAs (each designed to target a different, specific location in the genome) can be used for mutagenesis; after mutations have been induced, we select cells from the population based on our phenotype of interest; then, we sequence the selected cells’ genomes
Linking genetic variation with phenotypic variation
The purpose of QTL and GWAS is to identify genetic variations associated with complex traits. Some definitions:
- Linkage equilibrium: random association of alleles
- Linkage disequilibrium: non-random association of alleles; i.e. if the occurrence of alleles A and B in a haplotype are dependent on each other/non-random events
- Significance: makes finding linkage in human GWAS more likely (since local SNPs vary in correlation with other SNPs) and tell us where recombination hotspots are
- Higher linkage disequilibrium between alleles = higher likelihood of alleles staying together during crossing over
Genome-wide linkage maps
Linkage maps show the relative positions of genes/genetic markers along chromosomes.
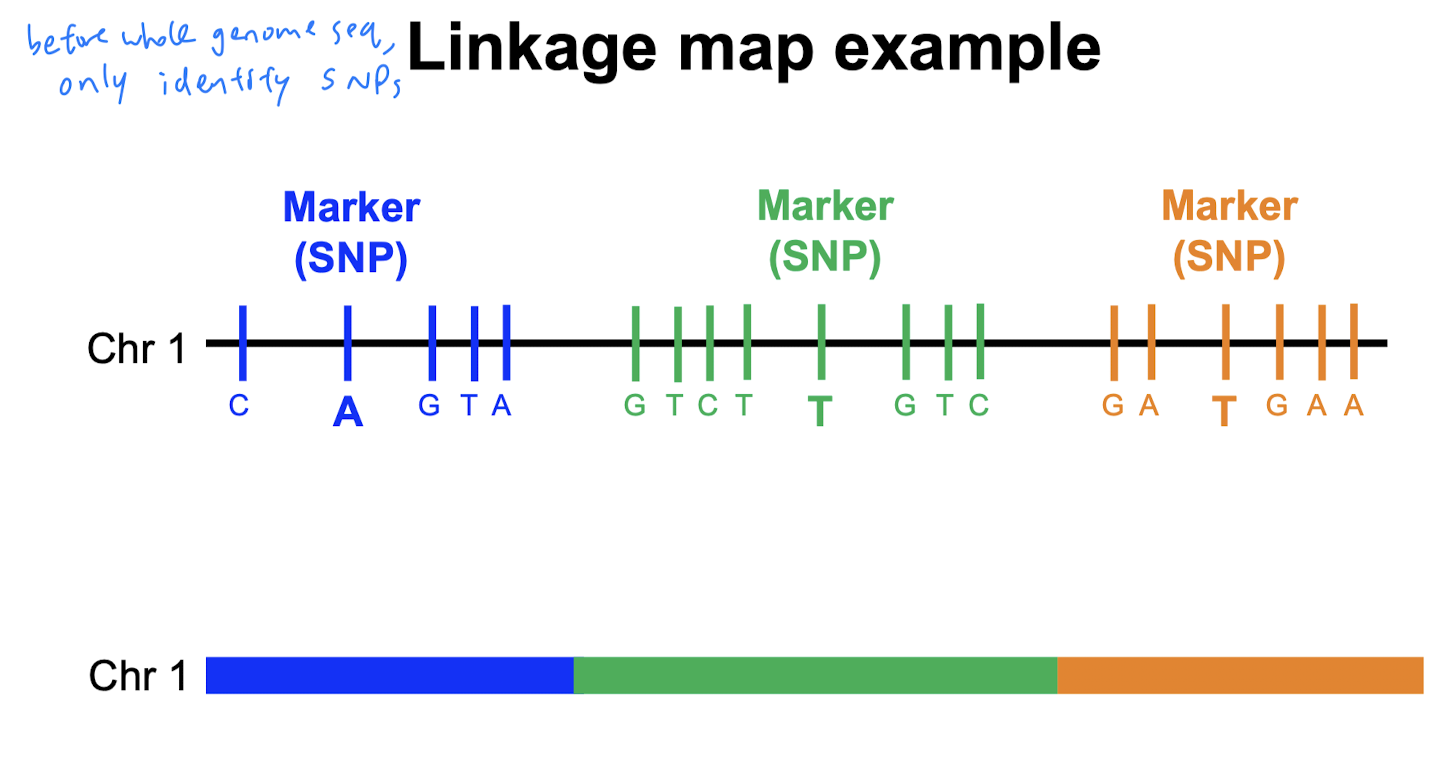 Before, we could sequence entire genomes, their full sequences were unknown. But, we had genome-wide linkage maps w/ molecular markers (e.g. SNPs - variations at a single nucleotide position in DNA among individuals)
Before, we could sequence entire genomes, their full sequences were unknown. But, we had genome-wide linkage maps w/ molecular markers (e.g. SNPs - variations at a single nucleotide position in DNA among individuals)
Genome-wide linkage maps were constructed based on the concept of linkage (tendency of genes close together on a chromosome to be inherited together). Scientists then measure recombination frequency during meiosis. Gene order and recombination frequencies are used to construct the linkage map. (no need to know the details).
Quantitative Trait Loci (QTL)
Definitions
- Quantitative genetics: genetic analysis of complex characteristics
- Quantitative trait (most traits in nature are quantitative)
- Continuous: varies continuously, falls on a distribution (e.g. human height)
- Meristic: measured in whole numbers (e.g. litter size)
- Threshold: measured by presence or absence (e.g. after accumulating a certain # of mutations)
- QTL: chromosome segments containing genes that influence a quantitative trait
- QTL mapping: locating genes that affect quantitative traits For quantitative traits, each genotype may produce a range of possible phenotypes. AA and Aa could both show blue eyes, for example, and we wouldn’t be able to tell what genotype someone is (AA vs. Aa) if they have blue eyes.
Steps
- Pick 2 parents, both homozygous, but homozygous for contrasting alleles at the same locus (e.g. AA and aa), so they have different phenotypes. Cross the 2 parents to produce F1 (all heterozygous).
- Interbreed F1s (both heterozygous so as to give us a spectrum of genotypes and phenotypes in F2) to create F2 progeny.
- Phenotype F2 for interested trait.
- Genotype each F2 for molecular markers (e.g. sequence SNPs throughout the genome).
- Test if any SNPs show linkage to interest trait:
- Create a linkage map using the genotyping data from F2.
- Analyze the genotypic data from the markers and phenotypic data from the traits. Goal: identify markers statistically associated with variations in the trait.
- Identify QTLs: regions of genome significantly associated with the trait variation are identified as QTLs
Genome-wide Association Studies (GWAS)
- GWAS def: a test of the association between SNPs across the genome (genotype) and a quantitative trait (phenotype)
- GWAS applications
- Provided new possible therapeutic targets to treat/cure disease
- Revealed new biology underlying important traits
- Found common alleles with large effects
Steps
- Collect DNA from many (often thousands of) cases (affected by disease) and controls (unaffected by disease)
- Genotype everyone for tagSNPs (minimal set of SNPs that tag most common haplotypes) throughout the genome
- Test for associations of each SNP genotype with disease status
QTL vs. GWAS
| Method | Study Group | Generations of Meioses Used | Probability of Finding Linkage | Genomic Resolution (once linkage found) |
|---|---|---|---|---|
| QTL | A cross or pedigree (more controlled breeding scheme) | A few (often 2 gens) | High (bc we control and see all phenotypes in our experiments) | Poor (Hundreds of kilobases to megabases of linkage disequilibrium regions) |
| GWAS | populations of affected and unaffected individuals | Thousands of historical meioses (bc looking at population) | Low (bc big population) | Great (~10 kilo bases of linkage disequilibrium regions) i.e. generates a tight linkage between marker and trait |
Developmental genomics
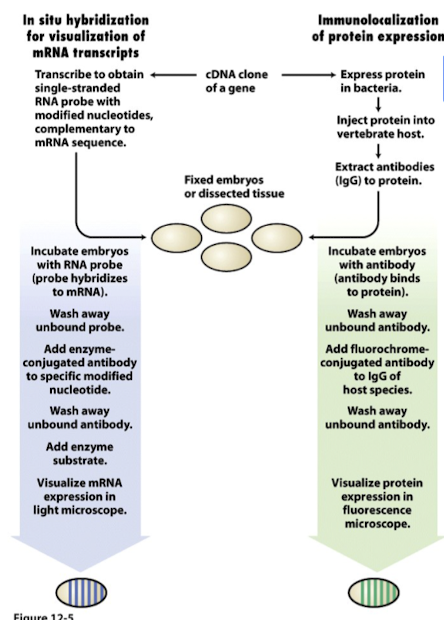
Visualizing gene and protein expression
In situ hybridization
- Purpose: visualize endogenous mRNA expression within tissue samples/embryos without the need for sequencing, using a single-stranded sequence-specific RNA probe
- I.e. can answer these questions
- Is a specific mRNA present (expressed)?
- If it’s present, where is it present?
- Anywhere we see precipitate present = is where mRNA is expressed (but remember, where mRNA is expressed =/= where protein is functional bc transcribed =/= translated) Steps
- Design a single-stranded probe specific (and complementary) to mRNA of interest: identify mRNA sequence to visualize, then design a single-stranded RNA probe complementary to our target mRNA sequence
- Make RNA probe via transcription: single-stranded RNA probe is made by transcribing our mRNA sequence of interest. During transcription, we incorporate UTPs (modified nucleotides), which are conjugated with haptens (molecules that can elicit an immune response and thus can be recognized by specific antibodies)
- Hybridize probe to target mRNA endogenously: incubate embryos with mRNA probe; probe will anneal to target mRNA present in sample bc probe and target mRNA have complementary sequences
- Wash away unbound probe
- Detect probe with an enzyme-conjugated antibody: use a specific antibody that recognizes the hapten conjugated to the probe; the antibody is conjugated to an enzyme, allowing for visualization of probe-target-mRNA complex.
- Wash away unbound antibody
- Add enzyme substrate
- Visualize mRNA expression in light microscope: enzyme attached to antibody catalyzes a reaction with a substrate. The enzyme degrades the tissue and forms a purple precipitate that coalesces around where the antibody is. Location and intensity of the precipitate indicate presence and quantity of target mRNA in sample.
Immunolocalization
- Purpose: visualize where a protein is expressed, using an antibody specific to our protein/transcription factor of interest (similar to ChIP-Seq)
- Answers these questions
- Is a specific protein/tf present? If so, where is it present? Steps
- Express protein of interest in foreign organism: produce protein/tf of interest in an organism different from the one being studied to ensure protein is recognized as foreign
- Inject protein of interest into host animal: host animal’s immune system will recognize protein as foreign and produce antibodies against it
- Extract and purify antibodies produced against protein: extract blood from immunized animal and purify the antibodies that specifically recognize the protein of interest
- Incubate embryos with antibody (antibody binds to protein): incubate purified antibodies with tissue/samples where we want to visualize protein expression. Anywhere protein is translated, the antibody will detect and bind to it.
- Wash away unbound antibody
- Add a secondary (used for detection and visualization) antibody: introduce a 2nd antibody that recognizes the primary antibody; 2nd antibody is conjugated to a detection marker (e.g. fluorescent dye or an enzyme catalyzing a colorimetric reaction)
- Visualize protein expression: use visualization technique (e.g. fluorescence microscopy) to detect signal from secondary antibody. Location and intensity of signal indicate presence and distribution of target protein within the sample.
Necessary vs. sufficient
How do we know if X (a gene, for e.g.) is necessary or sufficient?
- Necessary: without X, an effect does not occur
- Sufficient: with X, an effect occurs even when it shouldn’t
GAL4/UAS for ectopic expression
- GAL4/UAS is a tool to test sufficiency
- GAL4 is a transcription factor; GAL4 is introduced into an organism’s genome under the control of a tissue-specific promoter (so GAL4 will only be made in certain cells in certain tissues, depending on our chosen promoter)
- UAS is the enhancer that GAL4 binds onto; UAS is placed upstream (5’) of a gene of interest that we want to study/over-express, so the binding of GAL4 to UAS prompts the initiation of transcription of the downstream gene, leading to its expression
- Ectopic expression = express in an abnormal position (e.g. make eyes grow on legs)
How it works
- Design 2 separate genetic constructs
- GAL4 gene controlled by specific promoter that drives expression in desired tissue/cell type

- Gene of interest placed downstream (3’) of a UAS enhancer sequence (GAL4 binds to the UAS enhancer sequence) and a minimal promoter
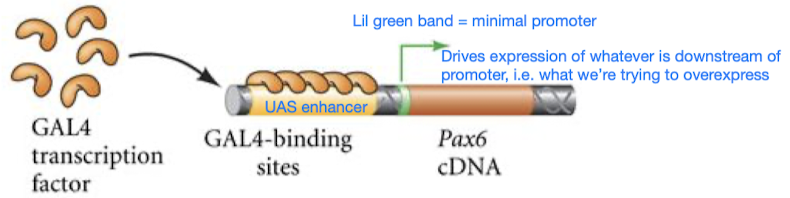
- GAL4 gene controlled by specific promoter that drives expression in desired tissue/cell type
- Introduce the 2 genetic constructs into host organism
- Tissue-specific GAL4 expression: the tissue-specific promoter drives the expression of GAL4 in desired cells/tissue
- UAS activation: GAL4 binds to the UAS sequences upstream of gene of interest in cells/tissues where UAS is expressed
- Gene of interest is expressed: the binding of GAL4 to UAS prompts the activation and expression of the downstream gene of interest (specifically in cells/tissues where GAL4 is expressed)
- Phenotypic analysis: organism is observed for effects of gene of interest’s expression in targeted cells/tissues. e.g. observe developmental outcomes
Outline
Lec 2 - Annotating protein-coding genes in the genome
- Purpose: bc genome sequence alone doesn’t tell you anything about the sequence’s function How to find protein-coding sequences in genomes?
- Find long open-reading frames (ORFs)
- Look for conserved sequences (comparative, evolutionary genomics)
- Coding sequences are highly conserved, esp among primates
- RNA-Seq
Review Session
DNA sequencing, assembly, and alignment
Building a genomic library
- see above
Forward genetics and complementation test
- Forward genetics: start with phenotype; interested in a phenotype, want to identify genes involved in that phenotype
- Mutagenic screen: mutagenize many random places in genome, screen for mutations that affect phenotypes of interest, generate homozygous flies for each mutant phenotype (to be able to do complementation tests)
- Complementation tests with mutants: we want to know if the mutations are in the same or separate genes
- If mutations are on separate genes, they will complement each other so progeny will have wildtype phenotype
- Complementation tests - things to know
- Are done with homozygous mutants
- Can tell us ho w many genes are involved in our phenotype of interest but not where the genes are located
- Mutations that complement each other = WT
- Mutations that dont’ complement each other = mutant phenotype
- Crossing mutants to WT flies show if a mutation is dominant or recessive over the WT allele
- Reverse genetics: start with genotype
QTL mapping
- More loci = more phenotypic classes
- Molecular markers: pre-determined regions in genome where there is known variation
- QTL alone won’t tell you which specific gene is associated with a phenotype; just will tell you the region that contains the gene
GWAS and linkage
- SNPs of interest have been separated by many thousands of meiosis events (vs. QTL only 2 generations, F1 and F2)
- So we need more molecular markers to identify loci of interest since there has been more separation among them
- LD vs. LE
- LE (linkage equilibrium): unlinked loci
- LD (linkage disequilibrium): linked loci; presence of an allele at one site can help predict what the allele at the second site will be
- LE vs. LD is on a spectrum, not black and white